Atoms, the fundamental building blocks of matter, present a fascinating, albeit complex, quantum system. Each atom comprises a positively charged nucleus surrounded by a cloud of negatively charged electrons, all governed by the laws of quantum mechanics. When atoms aggregate to form molecules, their interactions become intricate, posing significant challenges for simulation and computation. These challenges are not merely academic; they have profound implications for fields such as drug development and material design. Understanding how these atomic and molecular dynamics operate is crucial for innovations in solar cells, batteries, and pharmaceuticals.
The inherent complexity arises from the governing equations of quantum mechanics, notably the Schrödinger equation, which describes the energy states accessible to such systems. Solving these equations, particularly for large molecules, remains a computational nightmare. Even the most potent supercomputers may take days or even weeks to simulate molecular interactions involving just a few dozen atoms. When considering long time-scales essential for phenomena like protein folding or drug interactions, the computational demands can quickly spiral out of control.
Machine Learning: A Paradigm Shift in Molecular Simulation
Recent advancements in machine learning (ML) have ushered in a transformative approach to this age-old problem. Rather than laboriously solving the Schrödinger equation directly, researchers are now employing ML algorithms to predict the outcomes of electronic interactions within molecules reliably and efficiently. This shift has made it possible to bypass some of the heavy computational workload traditionally required for molecular dynamics simulations.
The essence of employing machine learning lies in its ability to recognize patterns and make predictions based on data, fundamentally changing how researchers approach molecular dynamics. Instead of separately modeling every variable, ML can efficiently learn the behaviors of electrons and their interactions from existing data. This not only expedites the simulation process but also empowers scientists to explore more complex systems than ever before.
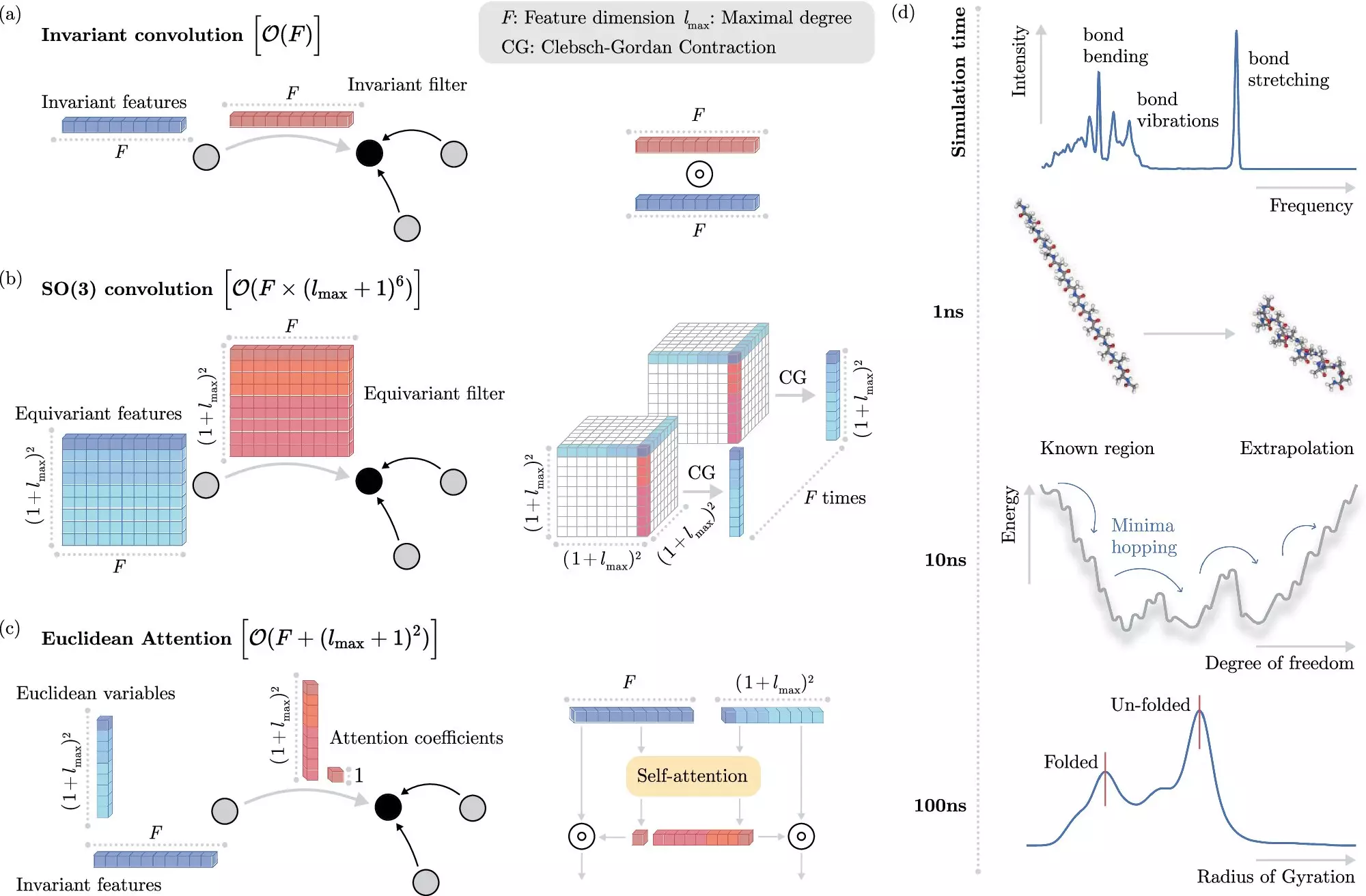
Nonetheless, the challenge of integrating quantum invariances—properties that remain unchanged regardless of the spatial configuration of the molecules—remains a barrier. Standard ML models often struggle with efficiency when attempting to account for these invariances, leading to protracted computation times.
The Breakthrough Algorithm from BIFOLD and Google DeepMind
In an exciting development, researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) in collaboration with Google DeepMind have created a revolutionary ML algorithm that focuses on streamlining the process of molecular dynamics simulation. This innovative approach separates the task of dealing with invariances from the core operations of the model. By adopting this method, the algorithm drastically minimizes the computational overhead ordinarily associated with these simulations, enabling them to run on a single computer node in mere days instead of months.
This leap in efficiency is nothing short of revolutionary for fields relying on molecular dynamics simulations. According to lead researcher Dr. Stefan Chmiela, the implications are vast. The new algorithm not only enhances our ability to simulate complex molecular systems but also opens doors for real-time insights into intricate atomic interactions. Such advancements could significantly influence the future of drug design, potentially eliminating the need for extensive trial-and-error laboratory work.
Pioneering Applications: A Glimpse into the Future
Demonstrating the capabilities of this new algorithm, the BIFOLD team has already made strides by identifying the most stable version of docosahexaenoic acid (DHA), an essential fatty acid crucial to human brain function. This type of complex analysis—previously considered unfeasible with traditional quantum mechanical approaches—illustrates the potential of the new ML model to perform scans of thousands of molecular candidates with remarkable precision and speed.
As Professor Klaus-Robert Müller points out, the combination of cutting-edge machine learning with physical principles not only addresses long-standing challenges in computational chemistry but also represents a paradigm shift toward simulating systems of larger scales and intricate interactions more accurately. This research marks a significant advancement not just for theoretical chemistry, but also in practical applications that hinge on our understanding of molecular dynamics.
The Path Ahead: Bridging Technology and Chemistry
The horizon for molecular simulation is bright, yet it emphasizes the need for continuous innovation. While current algorithms have made great strides, there is still substantial work to be done to accurately simulate larger, more complex systems, particularly those involving long-range physical interactions. The ongoing challenge lies in developing algorithms that maintain efficiency without compromising accuracy or detail.
As machine learning continues to mature, its integration into molecular dynamics could redefine the boundaries of research and application in chemistry and material science. From drug discovery that allows for faster responses to health crises, to breakthroughs in sustainable materials, the realm of possibility appears limitless. By harnessing these advanced techniques, we stand on the cusp of a significant scientific evolution that could ultimately transform our approach to solving real-world problems.
Leave a Reply